- Share
- Share on Facebook
- Share on LinkedIn
Genome editing method for E. coli based on CRISPR-Cas12
Cathy Bouchenot, Hans Geiselmann, Noël Scaramozzino, Alexandre Dawid, Stéphan Lacour, Delphine Ropers, Hidde de Jong
CRISPR technologies have become standard laboratory tools for genetic manipulation across all kingdoms of life. Despite their bacterial origins, the development of CRISPR tools for engineering bacteria has been slower than for eukaryotes; however, their function and application for genome engineering have been demonstrated in a variety of bacteria, and their adoption has become more widespread. CRISPR editing allows the deletion or insertion of DNA in vivo, either on the genome or on mobile elements such as plasmids.
We are implementing a simple plasmid-based system for genome editing (genome integration of small and large DNA fragments) in E. coli using a CRISPR-Cas12a system and a λ Red recombination system with a single cloning step (Jervis et al., Microbial Biotechnology, 2021).
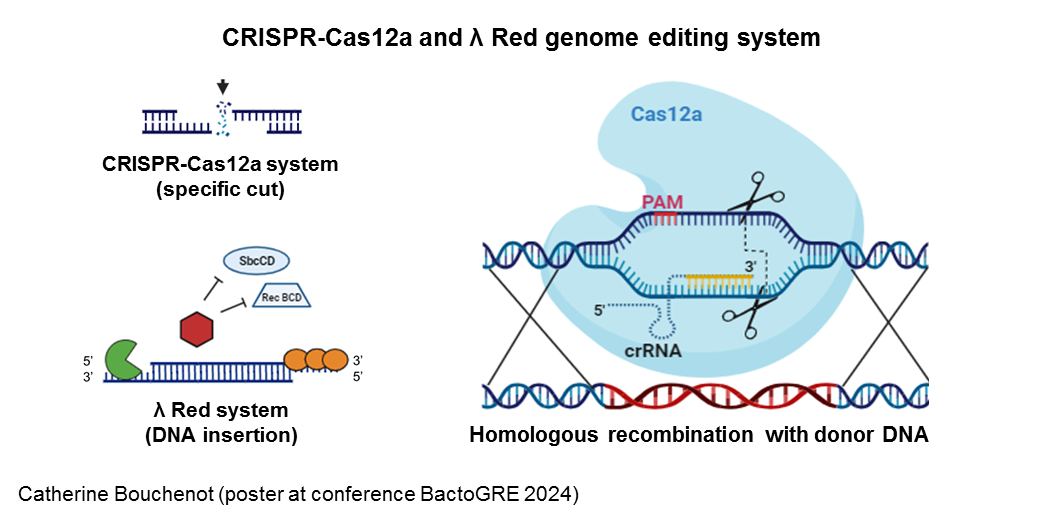
The CRISPR array is transcribed as a long precursor (pre-crRNA, not shown), which is then processed by cleavage in the repeat regions to produce individual target‐specific CRISPR RNAs (crRNA). These crRNAs direct the Cas12a nuclease to complementary double‐stranded DNA (dsDNA) targets, where the Cas12a nuclease creates a double-stranded break (DSB). The small guide RNAs (crRNA) can direct genome cleavage to any sequence adjacent to a 3 bp protospacer adjacent motif (PAM) by modifying the spacer region(s). Delivery of donor DNA (dsDNA), homologous to the cleavage site into recombination-compatible cells allows repair of the dsDNA break, replacing the target sequence with the dsDNA sequence in the process. Without repair, the cell dies, allowing a "live-dead" selection method.
- Share
- Share on Facebook
- Share on LinkedIn